Drawing a Circle to Multiply
A circle plot is a graph that looks similar a ring. Hither, I talk about how to draw a circle plot for genetic information with Python. The link to my lawmaking on GitHub is here.
The Data
In order to talk about the circle plot, allow'southward showtime talk well-nigh the data used for making the plot. I'm plotting differences in the cistron expression profiles of the three-spined stickleback. The three-spined stickleback is a pop model organism for studying population genetics, because they are the fish version of Darwin's finches. They have a broad range of morphology, are easy to continue in lab and discover in nature, and most importantly for this blog post, they tolerate salinity very well. In other words, they thrive in both fresh water and ocean water. I used RNA-sequencing data from two three-spined stickleback populations, one from fresh water and another from bounding main water.
When visualizing the data, I focused on 2 things: F-statistics ("Fst") and fold change in gene expression ("FC"). Fst is describes expected heterozygosity level, and information technology ranges betwixt zero and one. A low Fst value means there is less heterozygosity betwixt populations and they are genetically similar. A high Fst value means that the genetic differences between populations are pregnant. In summary, if the genetics of the two populations of fish were significantly different, the Fst would exist closer to i. If they were like, the Fst would exist closer to aught.
Fold modify describes whether one population expressed a gene more or less than the other population. The greater the absolute fold modify value, the greater the difference in the cistron's expression charge per unit between the two populations. In the data I used, fold change ranges from -ten,000 to ten,000, with a median around zero. For case, if the ocean water population expresses genes for regulating sodium level much more than the fresh water population, then the absolute fold change values for those genes would exist higher. If the two populations expressed the genes at the aforementioned level, the fold change values would be closer to zero.
Visualization
For visualization, I used Python and Pycairo, a Python library providing bindings for Cairo graphics.
Chromosomes to Arcs
In this circle plot, 21 chromosomes of the three-spined stickleback are represented as private arcs, and scaled according to their size, i.east. larger chromosomes are represented in larger arcs. The arcs are labeled with a roman number for their chromosome number. Each arc starts at base pair 0 and ends at the length of its chromosome. Base of operations pair locations are aggregated into buckets of 300,000 base pairs to go along the epitome legible. (Side notation, 3-spined stickleback has a genome that is 446 meg base pair long! Us humans have a 3 million base pair long genome.) Hither are the arcs for chromosomes I though IV:
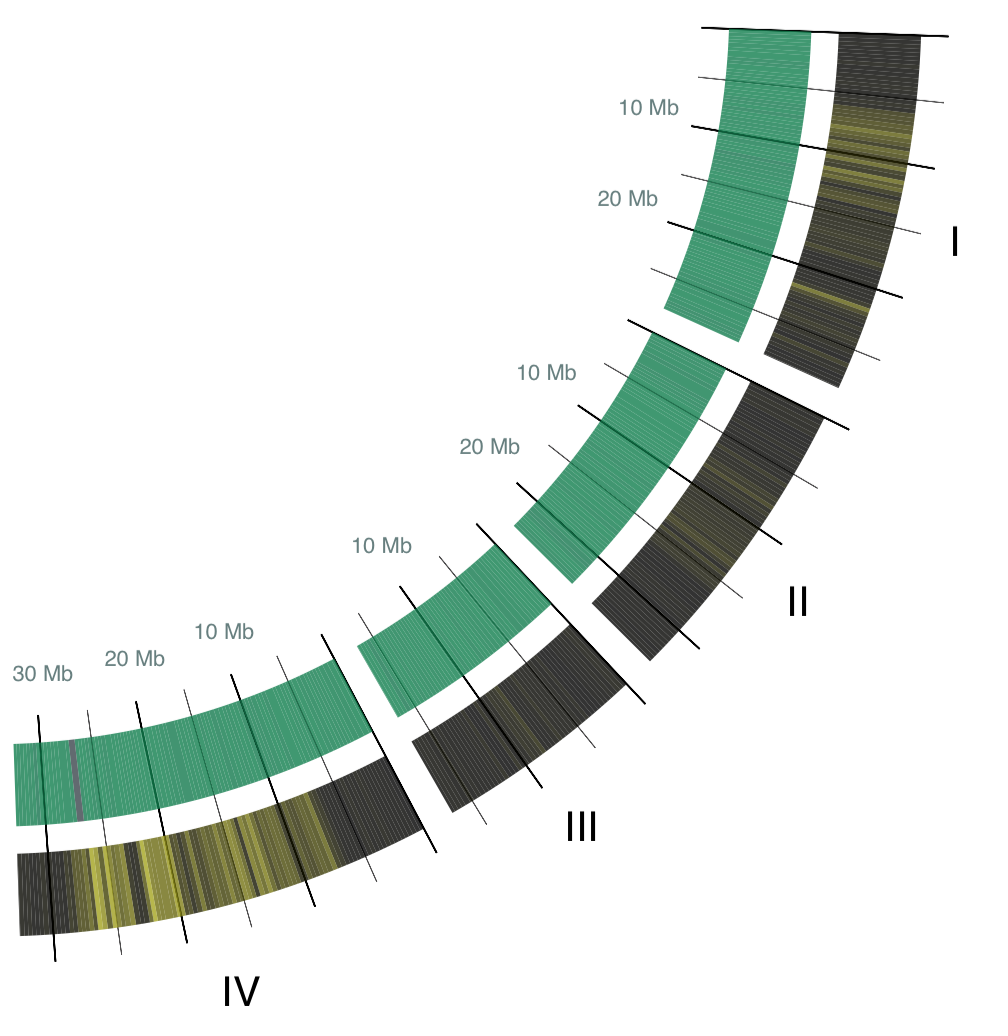
Statistics to Colors
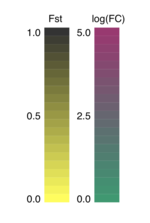
Translating Fst and fold change into colors was an interesting challenge. Equally Fst spans from goose egg to 1, it was relatively easy to choose a color based on the Fst value. A loftier Fst volition be colored in blackness, while low a Fst value will be colored xanthous. The code for generating an RGB colour value using Fst value is shown below:
if value_type == 'fst' : value = round ( float ( value ), 2 ) r = value chiliad = value b = 0 For fold change, I had to be a bit more creative. Fold modify values varied in a wide range, and I decided to transform fold change to log calibration, which ranges from -five to 5. And so I multiplied the values by 0.one so they would exist within -0.5 and 0.five. Then I took the absolute value of log(FC), because in this case, whether ocean or fresh water population overexpressed a gene is not an involvement, as long as we observe a fold change. A loftier fold change is shown in imperial, while a low fold change is shown in dark-green:
elif value_type == 'stat' : # Multiply past 0.1 so that the # values range from -0.5 to 0.five. value = round ( float ( value ) * 0.1 , 2 )) # Get the absolute value because fold modify is # subjective. It doesn't matter which population # expresses cistron more than as long as the difference is # apparent. value = abs ( value ) r = 0 + value g = 0.5 - value b = 0.3 This is the resulting legend:
The Resulting Plot
Later parsing the statistics files, assigning colors, and transforming chromosomes to arcs, here is the resulting circle plot: 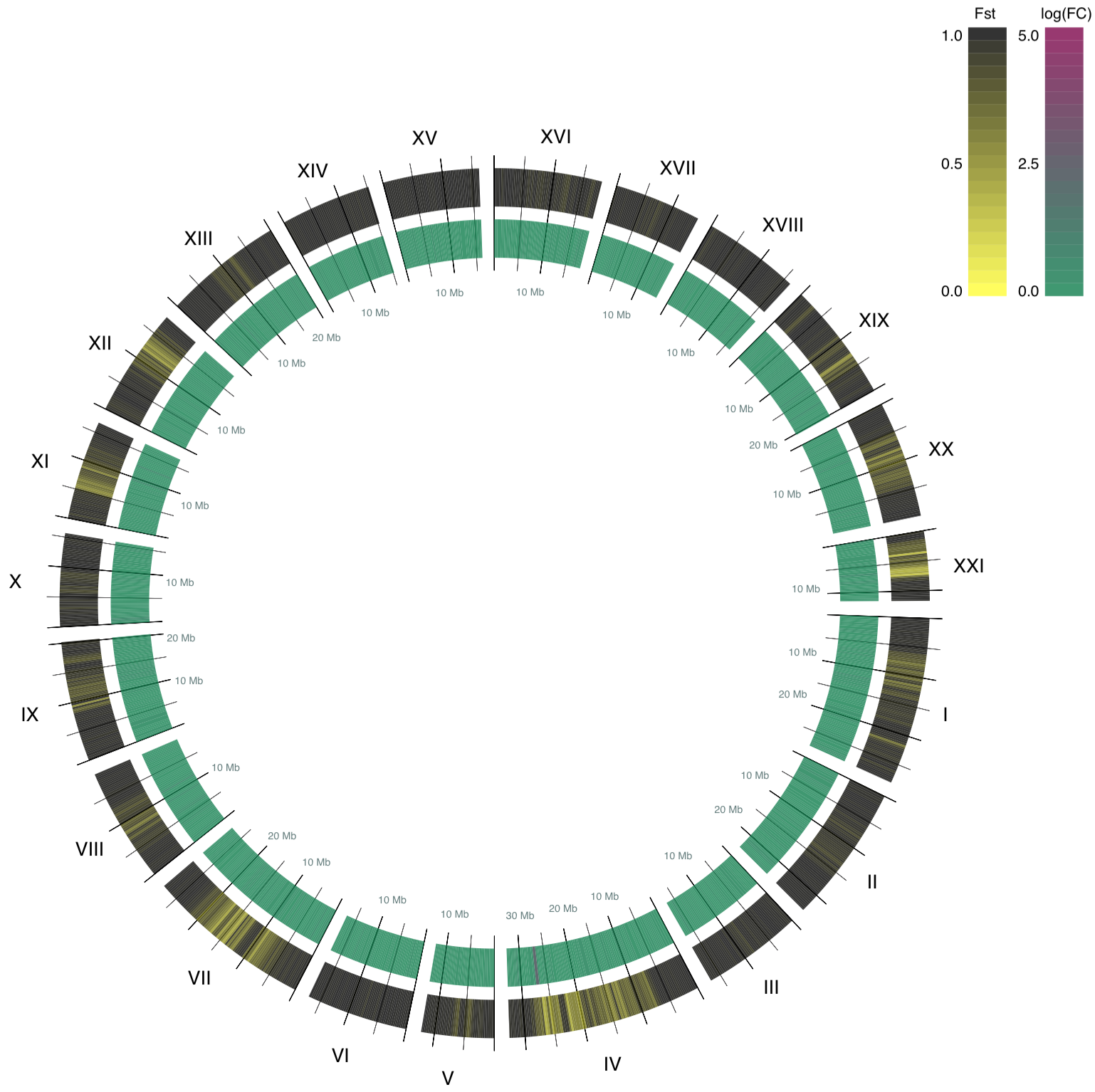
Conclusion
What conclusions can we describe from this visualization of the deviation between the genetic makeup of our fresh and ocean water populations?
Even though most of the genome seems to have college Fst values and therefore the genome is different between the populations, at that place are conserved regions in the genome (shown in yellowish color in this plot). Difference is interesting, but in biology, similarity in genomes often is more meaningful, because it implies that conserved regions contain important genes.
Information technology would be particularly interesting to dive further into chromosome IV because information technology contains a region that had a large fold changes, while it is highly conserved between the 2 populations. What genes have been protected from changes when the fish migrated between the dissimilar water types? Practice us humans take analogs for these genes? What would that hateful?
I am overall happy with how it turned out, and it was a great learning experience. Plus, information technology makes me realize the ability of visualization, and how it makes finding unique properties nearly the data then much easier. Give thanks you for reading.
Source: https://jeenalee.com/2016/02/25/circle-plot.html